Chk2 activation in response to double stranded DNA breaksThe best way to view the map is with on a PC with Firefox or Chrome and on a MAC with Safari or Chrome. 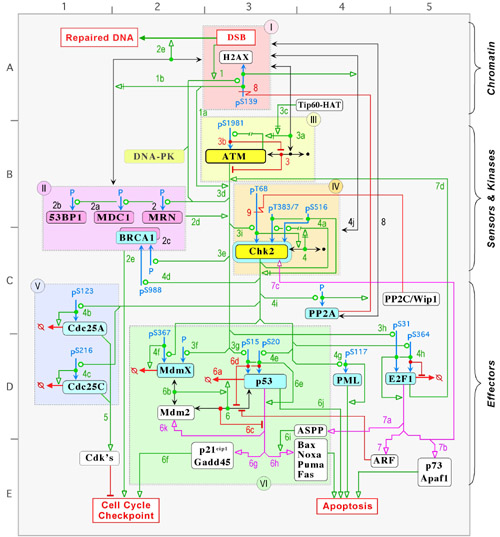
Yves Pommier, John N. Weinstein, Mirit I. Aladjem, and Kurt W. KohnImplemented by : Margot Sunshine, Hong Cao and David Kane DNA double-strand breaks (DSB) are among the most severe genomic lesions. Their repair requires cells to arrest cell cycle progression to avoid further damage during replication or transcription. DSB can be produced directly by ionizing radiation and certain anticancer drugs that bind to DNA (bleomycin, neocarcinostatin, topoisomerase II inhibitors such as etoposide or doxorubicin). DSB can also result from conversion of single-strand breaks by DNA polymerase collisions or �replication fork collapse� at sites of damaged DNA. These replication DSB occur normally in cancer cells, but their frequency is markedly enhanced by topoisomerase I inhibitors such as the camptothecin derivatives (topotecan and irinotecan) and DNA alkylating agents. Chk2, a highly conserved protein kinase, functions as a kinase relay for the PI3 kinase ATM, whose gene (Ataxia Telangiectasia Mutant) is mutated in Ataxia Telangiectasia patients. Chk2 activation leads to the activation of the G2 and S-phase cell cycle checkpoint, which is coupled with DNA repair. On an alternative pathway, Chk2 activation may also trigger p53 dependent apoptosis. This MIM underlines some remarkable features of the ATM-Chk2-p53 axis/network with its several positive and negative feedback loops, convergence of signals to achieve key molecular outputs, and the coupled ATM- and Chk2-mediated phosphorylation of p53 and other dual ATM-Chk2 substrates. A significant fraction of tumors and precancerous lesions overexpress activated Chk2. Therefore, Chk2 inhibitors might selectively act against such tumors. Because of the dual function of Chk2 (induction of p53-dependent apoptosis and cell cycle checkpoint activation coupled with DNA repair), it is plausible that a Chk2 inhibitor would also increase the therapeutic indices of DNA-targeted agents. In tumors defective for apoptosis (and p53), the Chk2 inhibitor would act primarily as a checkpoint inhibitor and prevent DNA repair, whereas in normal tissues (with functional p53) the Chk2 inhibitor would primarily block apoptosis and subsequent side effects. Map symbols Map Navigation |
|
This website is a development of the Genomics and Pharmacology Facility, Developmental Therapeutics Branch (DTB), Center for Cancer Research (CCR), National Cancer Institute (NCI). |
|
If you have questions or comments about this site, please send them to: webadmin@discover.nci.nih.gov. |
|
|
|
|
|
|